Mukoviszidose
Was ist das?
Die Mukoviszidose (Zystische Fibrose) ist eine schwere chronische Erkrankung. Sie stellt die häufigste angeborene Stoffwechselerkrankung der weißen Rasse dar. Es besteht eine Fehlfunktion aller schleimproduzierenden Drüsen im Körper. Es entsteht ein wasserarmes, sehr zähes Sekret, das entweder aus den Drüsen nicht abfließen kann und eine Entzündung hervorruft oder aber die ableitenden Gänge oder Bronchien verstopft und in ihrer Funktion beeinträchtigt. Daraus resultiert eine chronische Erkrankung vieler Organe, vor allem aber der Lunge und der Bauchspeicheldrüse. Gehäufte Lungenentzündungen, die Ausbildung von Bronchienaussackungen (Bronchiektasen), chronische Keimbesiedlung, Zerstörung des Lungengewebes, Sauerstoffmangel, Herzschäden, und eine gestörte Verdauung wegen der unzureichenden Freisetzung von Verdauungsenzymen sind die Folge. Aber auch der Darm, Leber, Galle, Nase, Nasennebenhöhlen sowie die Fortpflanzungsorgane können betroffen sein.
Ursache und Vorkommen:
Die Erkrankung wird durch Genveränderungen ausgelöst und ist erblich. Das Mukoviszidose-Gen (CFTR-Gen für cystic fibrosis transmembrane regulator) wurde erst 1989 entdeckt. Beide Elternteile müssen Anlageträger oder selbst erkrankt sein. Man muss im deutschsprachigen Raum mit einem CF-kranken Kind bei 2000-2500 Geburten rechnen.
Wie wird die Erkrankung entdeckt?
Es wird bisher kein Neugeborenenscreening durchgeführt. Sie Symptome sind sehr vielseitig: der sogenannter Mekoniumileus bei Neugeborenen, Mangelgedeihen, Durchfälle, häufige Bronchitiden und Lungenentzündungen, Entzündung von Nasennebenhöhlen, Salzhunger und vieles mehr.
Der erste diagnostische Schritt ist der so genannte Schweißtest, mit dem der Gehalt von Chlorid- und Natriumionen im Schweiß gemessen wird. Bei Mukoviszidose-patienten ist der Chlorid-Gehalt im Schweiß erhöht. Mithilfe einer Potentialdifferenzmessung an der Nasenschleimhaut oder einer Darmgewebeprobe kann im Krankheitsfall eine erhöhte Spannung nachgewiesen werden. Untersuchungen des Stuhlganges auf Fettgehalt und Verdauungsenzyme weisen ggf. die Störung der Funktion der Bauchspeicheldrüse nach. Genanalysen differenzieren dann die Genveränderungen. Lungenfunktionelle Untersuchungen, Röntgen- oder Computertomogrammaufnahmen des Brustkorbes, Sputum-untersuchungen auf Bakterien, Blutuntersuchungen und Funktionsanalysen der Bauchspeicheldrüse und der Leber werden in Abhängigkeit vom klinischen Verlauf durchgeführt.
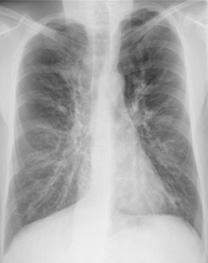
Abb1: Röntgenthoraxbild eines Mukoviszidosepatienten
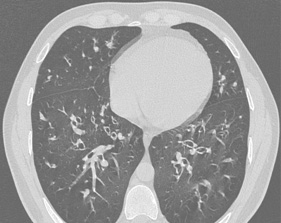
Abb2: Im CT des Thorax Nachweis von Bronchiektasen
Wie wird die Erkrankung behandelt?
Eine Heilung ist derzeit nicht möglich. Je früher und komplexer die Behandlung ist, desto besser ist die Lebenserwartung und Lebensqualität. Während früher die Kinder kaum das Erwachsenenalter erreicht haben, ist jetzt fast die Hälfte aller betreuten Patienten älter als 18 Jahre.
Für die Betreuung der Mukoviszidosepatienten ist ein interdisziplinäres Team aus spezialisierten Ärzten, Physiotherapeuten, Ernährungsberatern, Fachschwestern, Sozialberatern und Psychologen erforderlich. Ein CF-Zentrum erfüllt strenge Kriterien, die Erfahrungen mit dem Krankheitsbild voraussetzen und eine komplexe Betreuung der Patienten absichern.
Grundpfeiler der Behandlung sind die Sekretelimination mittels Krankengymnastik und speziellen apparativen Techniken, die Inhalationstherapie, die konsequente Therapie der Atemwegsinfektionen mit Antibiotika, die Behandlung der chronischen Atemwegsobstruktion, die ausreichende Vitamin –und Energie-Zufuhr und die Einnahme von Enzymen der Bauchspeicheldrüsen, um die normale Verdauung zu gewährleisten.
Neue Therapieansätze werden derzeit in klinischen Studien überprüft.
In fortgeschrittenen Fällen kommen die Sauerstofflangzeittherapie, die nichtinvasive Beatmung und die Lungentransplantation in Betracht.
Autorenteam
- Dr. med. Christian Franke
- Dr. Th. Heindl, Leipzig
- Dr. med. H.-P. Hlawa, Schmölln
- Dr. med. Dirk Koschel
- Dr. Looke, Suhl
- Dr. Christian Riedel, Bad Berka
- Dr. med. S. Riese
- PD Dr. Jens Schreiber
- Dr. Dagmar Täuscher
- Dr. Diane Wieczorek
- Dr. M. Wiemann, Magdeburg
- Dr. J. Winkler, Leipzig
- Dr. Susanne Ziebuhr